Zeitliche Orchestrierung von EU-HTA und nationaler Nutzenbewertung
Europäische Bewertung von Gesundheitstechnologien (EU-HTA)

Die Umsetzung dieser gemeinsamen klinischen Bewertungen wird schrittweise ab dem 12.01.2025 erfolgen und ist über einen längeren Zeitraum geplant: Beginnend mit onkologischen Medikamenten und Arzneimittel für neuartige Therapien (Advanced Therapy Medicinal Products, ATMPs) im Jahr 2025 bis zur gesamten Bandbreite an betroffenen Arzneimitten in 2030 (siehe Abbildung).
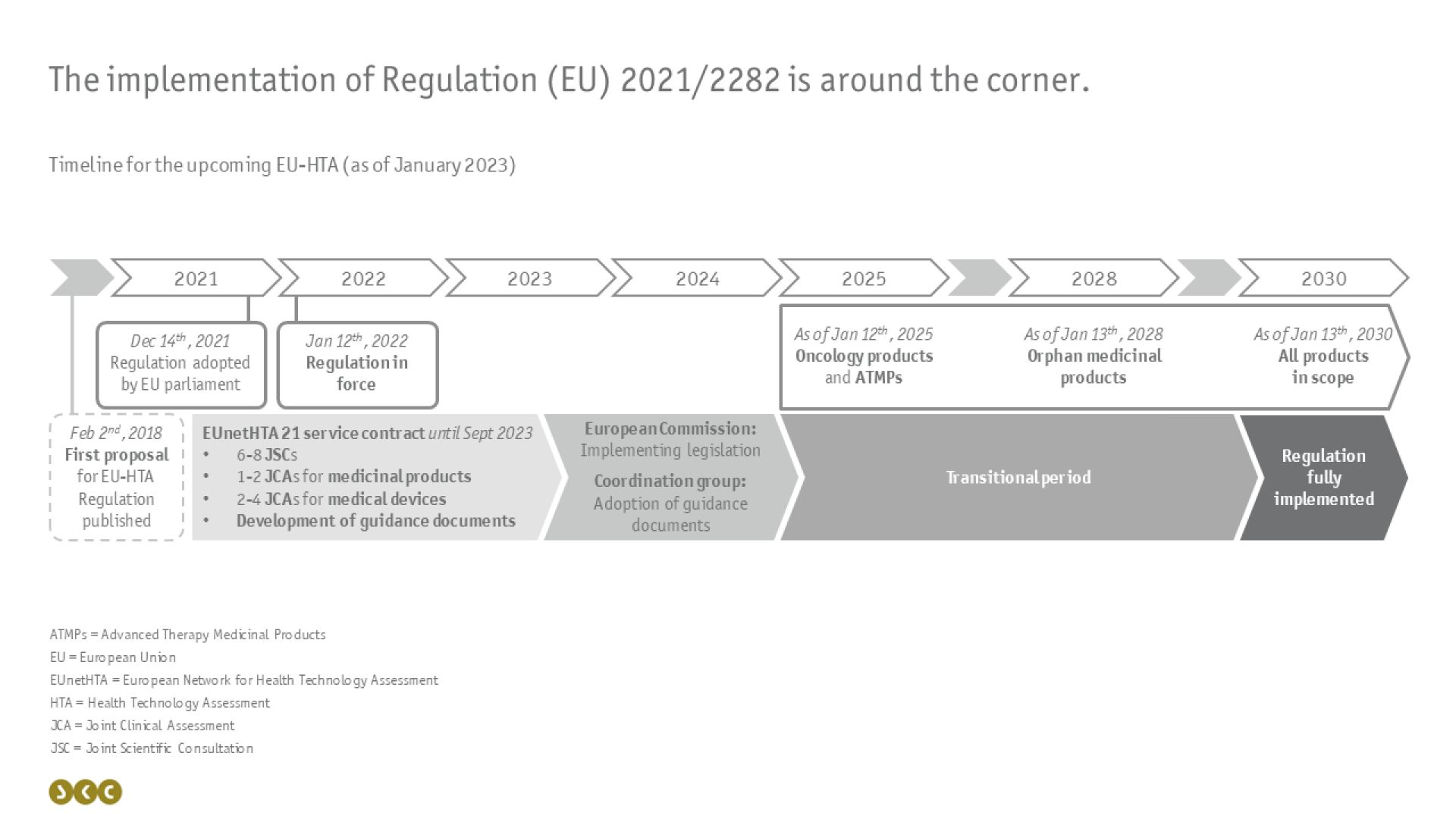
In der Zwischenzeit ist das Netzwerk EUnetHTA - ein Zusammenschluss verschiedener nationaler HTA-Behörden – damit beauftragt, Probedurchläufe für Beratungen und Bewertungen durchzuführen und die Verordnung ergänzende Leitlinien zu erstellen. Im Anschluss werden die erarbeiteten Dokumente und Erkenntnisse von der Europäischen Kommission in Durchführungsvorschriften überführt und von der Koordinierungsgruppe als Leitlinien übernommen.
Da die gemeinsame klinische Bewertung jedoch die nationalen HTA-Prozesse nicht vollständig ersetzen wird, ist es wichtig zu verstehen, wie die Abläufe auf europäischer und nationaler Ebene zeitlich ineinandergreifen werden (siehe folgende Abbildung).
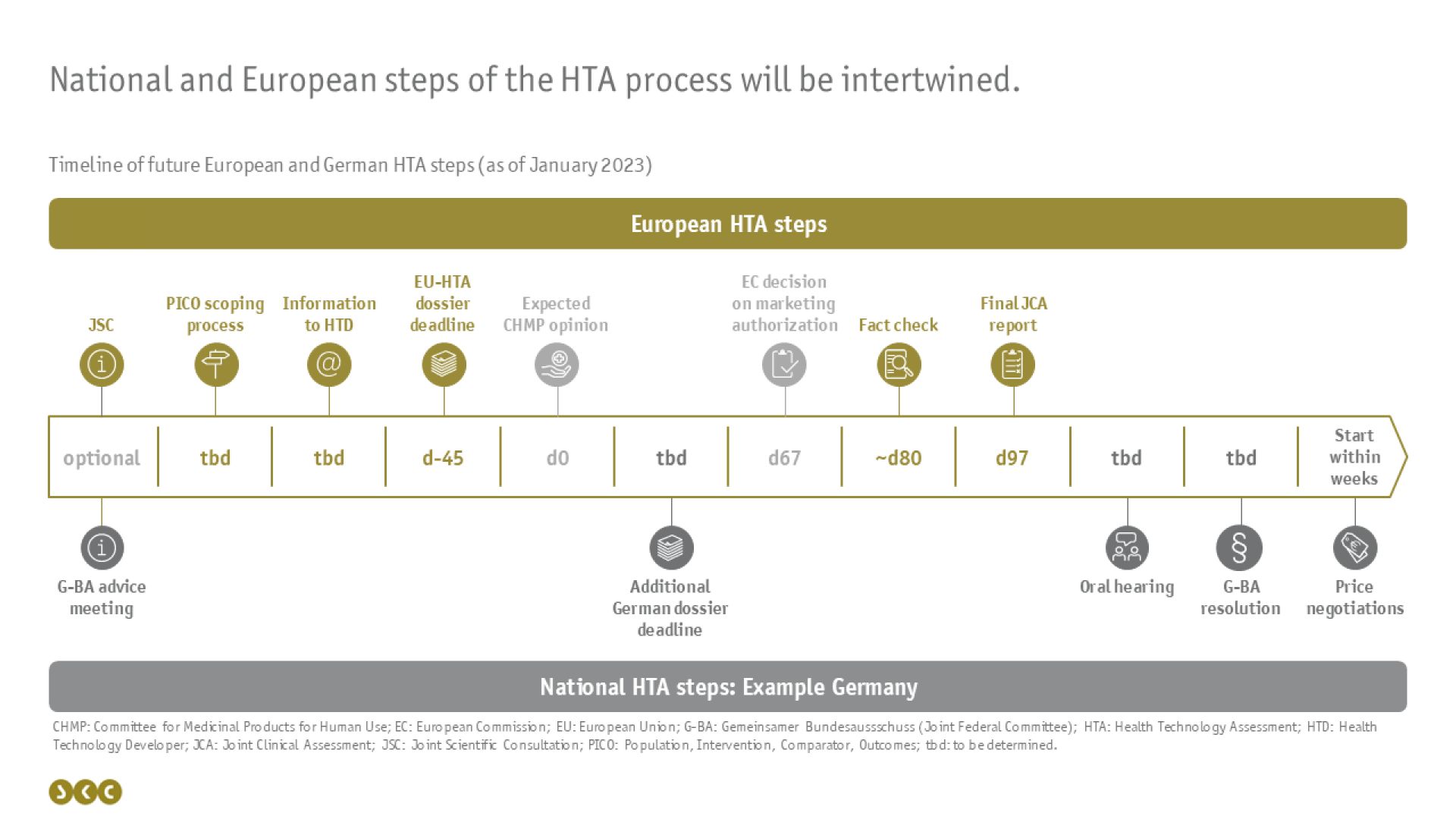
Zeitlicher Anker der JCAs auf europäischer Ebene wird das erwartete Datum der CHMP-Opinion sein: Bereits 45 Tage vorher muss das Dossier zur gemeinsamen klinischen Bewertung auf EU-Ebene eingereicht sein. Hierzu wird dem pharmazeutischen Unternehmer (pU) nach einer Befragung der einzelnen EU-Mitgliedsstaaten (Scoping process) mitgeteilt, welche PICO-Schemata in dem Dossier erfüllt werden müssen, sprich zu welchen Patientenpopulationen, Interventionen, Komparatoren und Endpunkten Ergebnisse vorgelegt werden müssen. Diese Mitteilung erfolgt voraussichtlich jedoch sehr kurzfristig (ggf. erst 55 Tage vor Einreichungsfrist des Dossiers), sodass dem pU nur wenig Zeit zur Erstellung des Dossiers bleibt. Entsprechend lohnt es sich, vorab ein freiwilliges Beratungsgespräch in Anspruch zu nehmen: entweder auf europäischer Ebene als JSC (Joint Scientific Consultation) oder separat auf nationaler Ebene in den EU-Mitgliedsstaaten.
30 Tage nach Zulassung des Arzneimittels wird der europäische JCA report – vergleichbar mit der Nutzenbewertung des IQWiG – veröffentlicht, nachdem der pU diesen einmal hinsichtlich faktischer Fehler kommentieren darf. Inhaltliche Anmerkungen oder Nachreichungen von Unterlagen wie sie aus der schriftlichen Stellungnahme des deutschen AMNOG-Prozesses bekannt sind, werden hierbei nicht möglich sein.
Der Beschluss über den Zusatznutzen des Arzneimittels sowie die Preisverhandlungen obliegen weiterhin den einzelnen Mitgliedsstaaten. Da sich in Deutschland bereits jetzt abzeichnet, dass nicht alle Informationen, die der gemeinsame Bundesausschuss (G-BA) für seinen Beschluss vorliegen haben möchte, in dem europäischen Dossier enthalten sein werden, wird mit an Sicherheit grenzender Wahrscheinlichkeit ein zusätzliches, ergänzendes Dossier für Deutschland erstellt werden müssen. Diese Möglichkeit besteht für alle Mitgliedsstaaten. Welche Länder davon Gebrauch machen werden und wann diese ergänzenden Module eingereicht werden müssen, steht noch nicht fest. Überhaupt ist es insgesamt noch unklar, wie die europäischen und nationalen Schritte des Verfahrens zeitlich ineinandergreifen werden. Ergänzend zu den vorgeschriebenen Schritten des HTA-Verfahrens, hat der G-BA bereits angekündigt, auch weiterhin eine mündliche Anhörung durchführen zu wollen.
Über neue Informationen halten wir Sie weiterhin auf dem Laufenden und stehen unseren Klienten beim strategischen Marktzugang ihres Arzneimittels oder Medizinproduktes sowohl unter aktueller nationaler als auch bei der zukünftigen geltenden europäischen Gesetzgebung gern unterstützend und beratend zur Seite.
Quellen:
